Pama-Nyungan verb classes
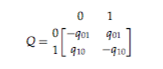
My current research project was born of my interest in the extension of modern computational methods to new areas of linguistic investigation. My dissertation provides an account of the evolution over time of verb conjugation classes in the Pama-Nyungan language family of Australia using Bayesian phylogenetic methods originally designed to model biological evolution. While the study of linguistic phylogenetics is relatively young, research has demonstrated the viability of the endeavor.
In terms of experimental design, I collected data about the verb conjugation classes of roughly 120 Pama-Nyungan languages, representing a sufficiently geographical and genealogical diverse sample of the family. Pama-Nyungan languages vary in terms of whether they contain or lack verb conjugation classes, how many classes they contain, and how verbs are divided into classes (i.e., based on valence or phonological properties of the stem). The properties of each given language give it a phylogenetic code or profile, which can then be used as input to Bayesian inference models in order to model the evolution of the verb conjugation class system within the context of the Pama-Nyungan family as a whole.
In addition to answering this inquiry into the nature and development of a specific typological feature, my research speaks to broader questions about linguistic phylogenetic methods. These broader questions, in turn, form the basis for future extensions of my work beyond the dissertation itself. The first of these is a more comprehensive look at the utility of linguistic trait reconstruction to limit hypotheses of the structure of phylogenies themselves. It is important to note that due to the statistical nature of Bayesian inference, current approaches to building these phylogenies that stretch beyond the historical record are not limited to the inference of a single family tree. Instead, a distribution of possible trees is generated, with the probability of a given tree or sub-tree (representing a subfamily in common terms) given as the relative frequency of its representation in the sample. Bringing in new typological data that was not used in the original phylogenetic inference allows us to test the various hypotheses that exist within the tree sample, and further hone our ability to constrain the sample itself.
Parker Brody
PhD Candidate
Yale University Department of Linguistics. Computational linguistics, Historical linguistics, Morphological Theory.